Terms
U, potential energy or chemical binding energy;
kb, Boltzmann's constant or 1.38041x10-23
T, absolute Temperature (Kelvin) where the temperature for the triple point of water is 273.15 Kelvin.
INTRODUCTION
The study of the growth of solid crystals, whether in a liquid solution or by vapor deposition in a vacuum chamber, is an important and intriguing process. It is a process which has many important uses in such industries as pharmaceuticals to semiconductors. The process for this project is simple. We start with a seed crystal or substrate with which atoms from the solution or vapor collide. Some of these atoms become a part of the crystal and the substrate grows. Many students have studied the growth of sugar crystals on a string placed in a supersaturated solution of sugar-water. A process of great importance today is the growth of near perfect crystals for the semiconductor industry. The computer industry is interested only in those processes which may consistently produce a well ordered crystalline solid, free of defects or vacancies. Amorphous solids, unstructured groups of atoms, are rejected. The production of crystals are determined by factors such as the quality and temperature of the seed crystal and the rate of growth.
We begin with a substrate of atoms, the seed crystal, and randomly drop atoms onto the substrate. Three things happen:
- the atom sticks at that site
- the atom bounces off the substrate completely
- the atom may move to a neighboring position on the surface of the substrate
The outcome is determined by the temperature and the potential (chemical binding) energy U of the atom on the surface of the substrate. Boltzmanns factor e(-U/kbT) determines the probability of sticking. Qualitatively we would expect the crystals to develop differently at different temperatures, at low temperatures the atoms tend to stick to the substrate and at high temperatures the atoms' thermal energy allow them to bounce off the substrate.
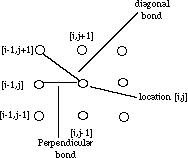
Figure 1. At location [i,j], there are eight neighbors. The atom at [i,j] would have two types of bonds: a bond with a neighbor in the same row or column is a perpendicular bond, while the bonds with the four remaining neighbors are called diagonal bonds.
THE PROJECT
This procedure is accomplished for each of the four perpendicular neighbors of the site, top, right, bottom, and left. (Here, let j=1 be our present position, j=2: top, j=3:right, j=4:bottom, j=5:1eft). The five values of the PE's are summed to calculate a total potential energy for that site, PETOTAL. Now for each of the five sites a probability factor is calculated by dividing the individual energy values by the total: PROBj=PEj/PETOTAL. Next the simulation calculates a random number between zero and one, RAND, against which we will compare the probability of current position:
- if ProB 1 >= RAND we record a 1 at location [i,j] and the "clock" value at time[i,j];
- else if PROB 1 + PROB2 > RAND let [i,j+l], the top, be our current position and go tbrough the whole process again;
- else if PROB 1 + PROB2 + PROB3 > RAND let [i+l,j], the right, be our current position and go through the whole process again;
- else if PROB l+PROB2+PROB3+PROB4>RAND let [i,j-l ], the bottom, be our current position and go through the whole process again;
- else let [i-1,j], the left, be our current position.
This process gives the atom the ability to jump to different sites and calculate whether the atom might stick at a site which borders the selected site. The procedures exist if:
- ST = 0 and DI = O (there are no neighbors)
or
- the column chosen is either the first or last colutnn, or the row selected is at the top of the grid definition, the top row.
Whether or not the atom sticks, the programs should increment the 'clock' and go on to find the next random column. The clock for this program will be an interation count through the routine. The clock will be initialized to zero at the beginning of the program. The time required to fill up the grid with crystals depends on the Boltzmann temperature constant and tbe bonding strengths. For a 75x30 grid a thousand clock cycles should be sufficient. Due to system and time constraints only results from some of the clock cycles will be saved. The user will determine which clock cycles to save. In order for the user to choose, they should be prompted for:
- the name of the output file
- the minimum clock value at which to start saving the data
- the maximum clock value at which time the algorithm terminates
- the number of clock cycles skipped to the next data saved, this is done by using the Pascal modulus operator, 'mod'
Psuedo-program:
Main program:
initialize; {call the initialize procedure}
get_parameters; {call the procedure which asks the user for the constant values}
loop: while clock <= maximum time
random_column:
{get a random column and find the row just above the
substrate}
monte_carlo(thiscolumn, thisrow); {pass the column and row
of the algorithm}
if: the time checks are correct
output the data in AVS format;
endif;
increment clock;
endwhile;
Initialize:
clock = 0;
all the entries for location[i,j] will be zero except for the bottom row, location[i, 1] will
Get parameters:
prompt user for the parameters kbT, diagonal bond strength, perpendicular bond strength, minimum and maximum clock cycles, and the number of clock cycles to skip before saving the data, and the name of the output file.
Random Column:
get a random integer value between 1 and the maximum column; drop from the top to the bottom to find the row just above the substrate below, this includes the diagonal neghbors, also;
Monte Carlo procedure:
we need the arrays of reals: BP[1..5], PE[1..5], PROB[1..5]
Sticking calculation procedure (i, j, k):
if location [i, j] = 1
BP[k] = PE[k] = 0.0;
else
calculate the number of diagonal neighbors and
perpendicular neighbors; calculate BP[k], PE[k] and
PETOTAL as discussed earlier;
[it is important to keep in mind that not every location
has eight neighbors]
initialize BP[ ] and PE [ ] to zero;
call sticking (this column, thisrow, neighborlocation);
conditions:
if there are no neighbors
exit routine;
else if the column is the last or first column or the top row
exit;
else
pass the up (neighbor location = 2), right (nl=3), bottom
(nl = 4), and left (nl = 5) column, row, and "nl" values to
sticking procedure.
calculate a random value, RAND, between 0 and 1;
check the random comparisons as discussed earlier,
updating the crystal information when necessary
otherwise allow the atom to jump to its perpendicular
neighbor.
Output procedure:
for each time iteration saved, there are two output files - an AVS header file and the data file.
DISCUSSION
With the flexibility of the user prompted parameters we may investigate the various properties of the crystal growth process. Figures 2 & 3 show some of the different crystals which develop under different conditions.

Figure 2. kT=28, diagonal bond = 100, perpendicular bond =10

Figure 3. kT=5, diagonal bond=100, perpendicular bond=10
ANIMATIONS
SI 1998
ACKNOWLEDGEMENTS
This code was developed from a simulation developed by Berkowitz, S.J., and Haase, D.G., and was originally written in BASICA for the IBM PC.
Leslie Southern is the OSC coordinator for the Crystal Growth project. Leslie's office is in 420-3. Please contact Leslie to set up appointment(s) for consultation.
For assistance, write si-contact@osc.edu or call 614-292-0890.